|
3. Catalysis: Olefin Polymerization,
Oscillating Chemical Reactions, and Steroid Oxidation
Methods: Experimentation, Catalysis, Auto-Catalysis, Dative Bonding,
Halogen Bonding, Oscillating Chemical Reactions, Kinetics Measurements,
Kinetics Simulations, Ab Initio Theory, DFT Theory, Multi-Layer Methods
(ONIOM), Thermodynamics.
3.1. Transition Metal-catalyzed, MAO-assisted Olefin Polymerization.
The low pressure polymerization of α-olefines with catalysts that combined
aluminum alkyls and transition metal complexes (i.e., Ti, Zr) was discovered
more than half a century ago, and the significance of the Ziegler-Natta
polymerization has steadily increased ever since. Methylaluminoxane (MAO)
is usually employed as co-catalyst. A major advance occurred in
1990's with reports by the groups of Brookhart and Gibson that described
first examples of a new generation of homogeneous catalysts for ethylene
polymerization. The precatalysts are neutral Fe(II) and Co(II) complexes
formed by addition of tridentate pyridine bis-imine ligands to the appropriate
metal salt. The precatalysts are employed in nonpolar organic solvents
in the presence of a very large excess (100-1000 equiv.) of MAO as
co-catalyst. The iron catalysts showed better activities in both studies
and their performance parameters were comparable to the most active
Ziegler-Natta catalysts. More recently, Sun and coworkers of the Chinese
Academy of Sciences, Beijing, explored structurally similar bidentate
bis(imino)pyridyl Fe(II) complexes, tridentate 2,8-bis(imino)quinolines
Fe(II) complexes, and related systems with nickel and titanium also have
been studied.
While the organometallic precatalysts are well characterized, very
little is known about the structure(s) and the function(s) of the active
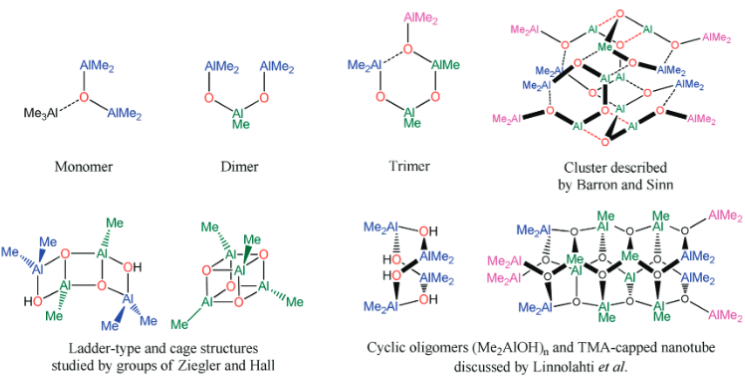
MAO species. Methylaluminoxane (MAO) is a generic term used to describe
the products of 'controlled' hydrolysis of trimethyaluminum (TMA,
Me3Al)
and modified methylaluminoxane (MMAO) is obtained by hydrolysis of TMA with
admixtures of other trialkylaluminum compounds (e.g., tBu3Al).
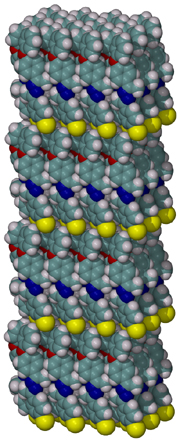
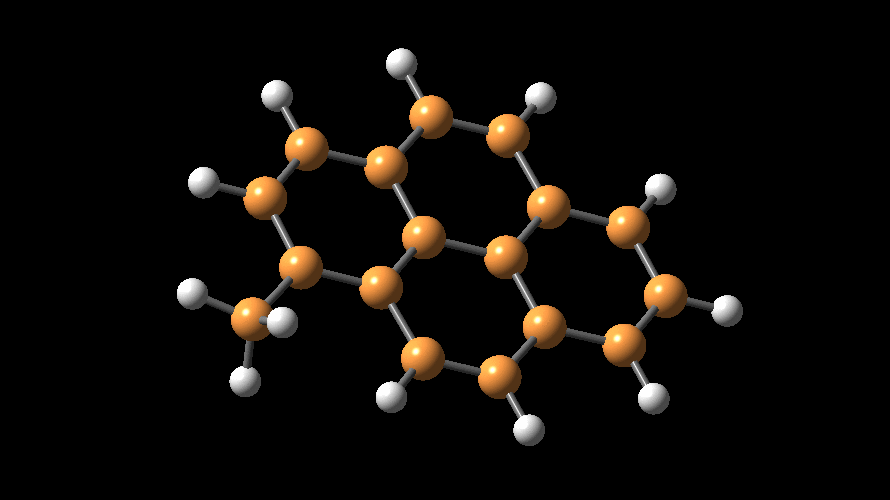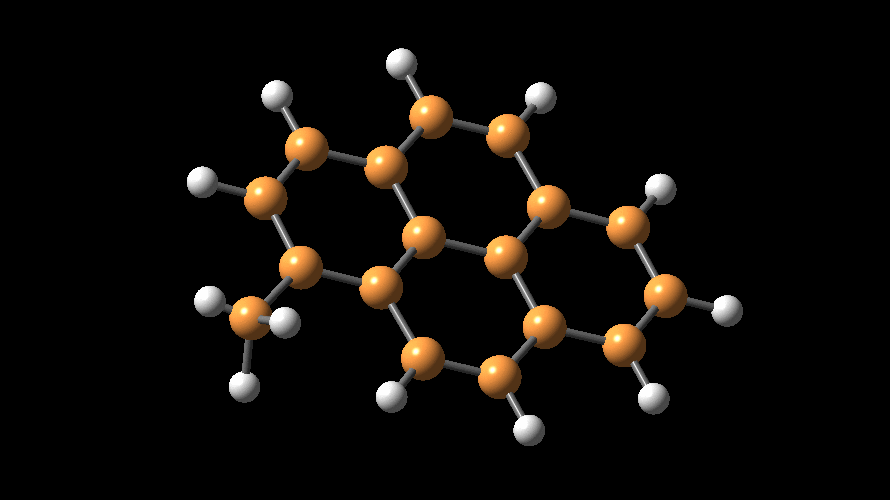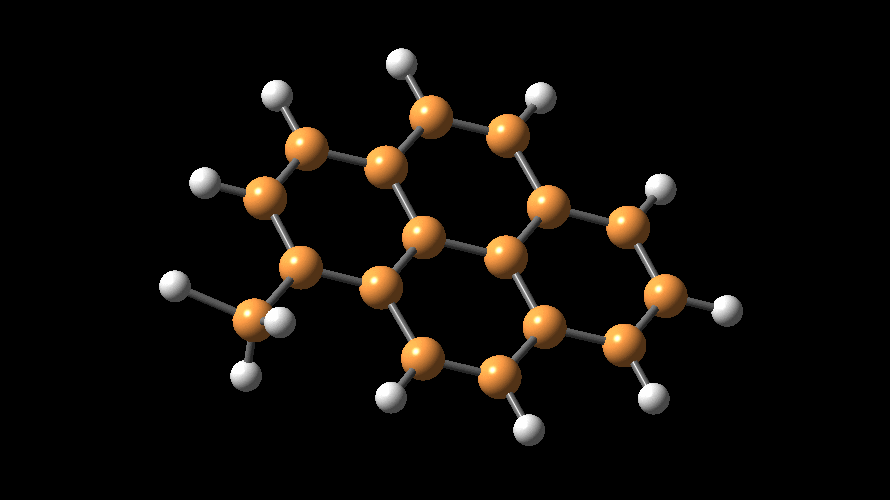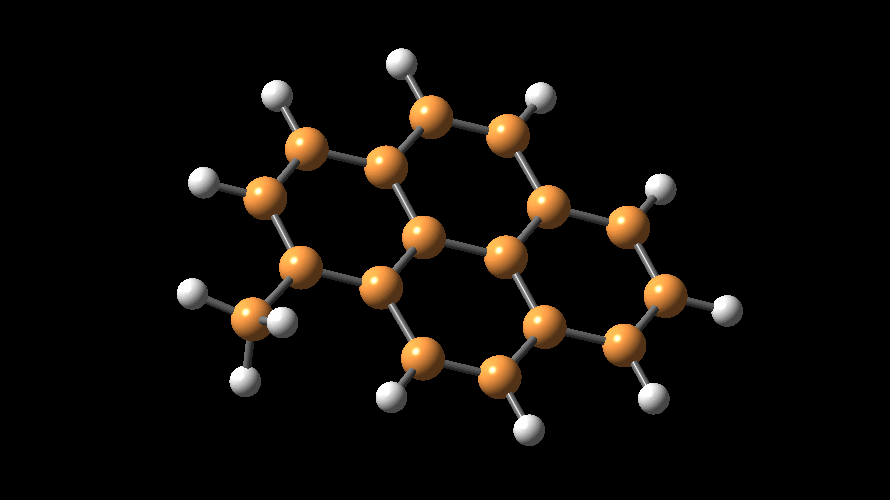
The compositions of MAO and MMAO are unknown, they depend on their formation
processes, and a variety of species have been discussed (see Figure).
It is our hypothesis that cyclic aluminoxanes are formed by partial hydrolysis
of TMA and that these cyclic aluminoxanes are involved in the catalysis.
The O-atoms in cyclic aluminoxanes are superior Lewis donors and much
more prone to coordinate to iron as compared to acyclic MAO species.
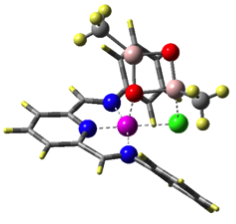
Hence, we are studying the olefin polymerization by MAO-ligated Fe-catalysts
(see Figure).
|
|